Tutorial pokazuje praktyczny sposób uruchamiania obliczeń Quantum ESPRESSO w systemie kolejkowym SLURM na klastrze Tryton Plus.
Pierwszy krok: Obliczenia SCF
W pierwszym kroku wykonujemy obliczenia SCF (Self-Consistent Field) za pomocą programu pw.x. Celem tego etapu jest wyznaczenie samouzgodnionego potencjału Kohn–Shama oraz gęstości elektronowej dla grafenu.
Wynikiem obliczeń SCF jest katalog ./tmp/gr.save, zawierający m.in.: gęstość ładunku, potencjał efektywny, funkcje falowe oraz metadane struktury. Ten katalog jest niezbędny dla wszystkich kolejnych kroków i stanowi punkt odniesienia dla obliczeń pasmowych.
SCF uruchamiamy na dwóch węzłach z równoległością po punktach k (-npool 24), co zapewnia dobrą skalowalność dla bardzo gęstej siatki k-punktów.
Plik gr_scf.in
&CONTROL calculation = 'scf' prefix = 'gr' outdir = './tmp/' tstress = .true. tprnfor = .true. / &SYSTEM ibrav = 4 A = 2.4560 C = 20.0 nat = 2 ntyp = 1 ecutwfc = 80.0 ecutrho = 500.0 nbnd = 120 occupations = 'smearing' smearing = 'mv' degauss = 0.02 / &ELECTRONS conv_thr = 1.0d-12 mixing_beta = 0.2 diagonalization = 'david' / ATOMIC_SPECIES C 12.011 C.pbe-n-kjpaw_psl.0.1.UPF ATOMIC_POSITIONS crystal C 0.0 0.0 0.25 C 0.3333333333 0.6666666667 0.25 K_POINTS automatic 96 96 1 0 0 0
Plik wsadowy do systemu kolejkowego SLURM 1_gr_scf.slurm
#!/bin/bash #SBATCH -J qe-scf #SBATCH -p test #SBATCH -N 2 #SBATCH --ntasks-per-node=48 #SBATCH --time=00:06:00 module load trytonp/quantum_espresso export OMP_NUM_THREADS=1 set -euo pipefail srun --mpi=pmi2 pw.x -npool 24 -ndiag 2 -i gr_scf.in > gr_scf.out
Używamy 2 węzłów z 48 zadaniami na węzeł, co daje łącznie 96 zadań. Podział na pule procesorów jest ustawiony na 24 pule (-npool 24), a liczba procesów diagonalizacji na 2 (-ndiag 2).
Uruchomienie obliczeń:
sbatch 1_gr_scf.slurm
Drugi krok: Obliczenia BANDS (pw.x)
W drugim kroku wykonujemy obliczenia pasm energetycznych (calculation = 'bands') również przy użyciu pw.x. Ten etap nie jest już obliczeniem samouzgodnionym – program korzysta z potencjału i gęstości wyznaczonych w kroku SCF.
Obliczenia prowadzone są wzdłuż zadanej ścieżki wysokosymetrycznej w przestrzeni odwrotnej (K_POINTS crystal_b), typowej dla struktury grafenu (Γ–M–K–Γ). Dla każdego punktu k diagonalizowany jest hamiltonian Kohn–Shama, co daje energie pasm.
Ten krok jest obliczeniowo kosztowny (duża liczba pasm i punktów k), dlatego stosujemy kontrolowaną równoległość: ograniczoną liczbę procesów MPI (-n 24) oraz podział po k-punktach (-npool 6), co w praktyce daje najlepszy kompromis między wydajnością a stabilnością.
Wynikiem tego etapu jest uzupełniony katalog ./tmp/gr.save, zawierający funkcje falowe dla całej ścieżki k, które zostaną użyte w postprocessingu.
Plik wsadowy gr_bands.in
&CONTROL calculation = 'bands' prefix = 'gr' outdir = './tmp/' / &SYSTEM ibrav = 4 A = 2.4560 C = 20.0 nat = 2 ntyp = 1 ecutwfc = 80.0 ecutrho = 500.0 nbnd = 120 occupations = 'fixed' / &ELECTRONS conv_thr = 1.0d-10 startingwfc = 'file' diagonalization = 'cg' diago_thr_init = 1.0d-5 diago_cg_maxiter = 200 / ATOMIC_SPECIES C 12.011 C.pbe-n-kjpaw_psl.0.1.UPF ATOMIC_POSITIONS crystal C 0.0 0.0 0.25 C 0.3333333333 0.6666666667 0.25 K_POINTS crystal_b 4 0.000000 0.000000 0.000000 60 0.500000 0.000000 0.000000 60 0.333333 0.333333 0.000000 60 0.000000 0.000000 0.000000 1
Plik wsadowy do systemu kolejkowego SLURM 2_gr_bands.slurm:
#!/bin/bash #SBATCH -J ge-bands #SBATCH -p test #SBATCH -N 1 #SBATCH --ntasks-per-node=48 #SBATCH --time=00:06:00 module load trytonp/quantum_espresso export OMP_NUM_THREADS=1 set -euo pipefail srun --mpi=pmi2 -n 24 pw.x -npool 6 -ndiag 2 -i gr_bands.in > gr_bands.out
Używamy 1 węzła z 48 zadaniami na węzeł, co daje łącznie 48 zadań. Podział na pule procesorów jest ustawiony na 6 puli (-npool 6), a liczba procesów diagonalizacji na 2 (-ndiag 2).
Uruchomienie obliczeń:
sbatch 2_gr_bands.slurm
Trzeci krok: Postprocessing bands.x
W trzecim kroku uruchamiamy program bands.x, który wykonuje postprocessing wyników obliczeń BANDS. Program ten nie liczy już hamiltonianu, lecz odczytuje wcześniej zapisane funkcje falowe i generuje dane gotowe do wizualizacji struktury pasmowej.
Etap ten jest szczególnie intensywny operacyjnie (I/O), ponieważ wymaga wielokrotnego odczytu dużej liczby plików z katalogu gr.save. Aby zminimalizować czas dostępu do danych, katalog ./tmp/gr.save jest kopiowany do lokalnego systemu plików w RAM (/tmp) na węźle obliczeniowym.
Takie podejście niesie ze sobą pewne ograniczenia: można wykorzystać tylko jeden węzeł obliczeniowy, bo /tmp jest lokalny dla każdego węzła – inne węzły nie mają do niego dostępu.
Następnie bands.x uruchamiany jest na jednym węźle z ograniczoną liczbą procesów MPI (-n 24), co w praktyce daje znaczące skrócenie czasu wykonania w porównaniu do pracy na współdzielonym systemie plików.
Wynikiem tego kroku są pliki gr_bands.dat oraz gr_bands.dat.gnu, które można bezpośrednio wykorzystać do tworzenia wykresów struktury pasmowej grafenu.
Plik wsadowy do systemu kolejkowego SLURM 3_gr_bands_pp.slurm
#!/bin/bash
#SBATCH -J ge-bands-pp
#SBATCH -p test
#SBATCH -N 1
#SBATCH --ntasks-per-node=48
#SBATCH --time=00:03:00
module load trytonp/quantum_espresso
export OMP_NUM_THREADS=1
set -euo pipefail
echo "JOBID=$SLURM_JOB_ID HOST=$(hostname)"
echo "PWD=$SLURM_SUBMIT_DIR"
cd "$SLURM_SUBMIT_DIR"
if [ ! -d ./tmp/gr.save ]; then
echo "ERROR: ./tmp/gr.save not found. Run SCF+BANDS first." >&2
exit 2
fi
# ramdisk na tym nodzie
SHM="/tmp/qe_gr_${SLURM_JOB_ID}"
rm -rf "$SHM"
mkdir -p "$SHM"
echo "Copying gr.save to $SHM ..."
cp -a ./tmp/gr.save "$SHM/"
# generujemy input do bands.x wskazujący na ramdisk
cat > gr_bands_pp_shm.in <<EOF_INNER
&BANDS
prefix = 'gr'
outdir = '${SHM}/'
filband = 'gr_bands.dat'
/
EOF_INNER
echo "Running bands.x (n=24) ..."
srun --mpi=pmi2 -n 24 bands.x < gr_bands_pp_shm.in > gr_bands_pp.out
# kopiujemy wyniki na trwały FS
cp -a "${SHM}/gr_bands.dat"* . 2>/dev/null || true
# sprzątanie shm
rm -rf "$SHM"
echo "bands.x done: $(date -Is)"
Uruchomienie obliczeń:
sbatch 3_gr_bands_pp.slurm
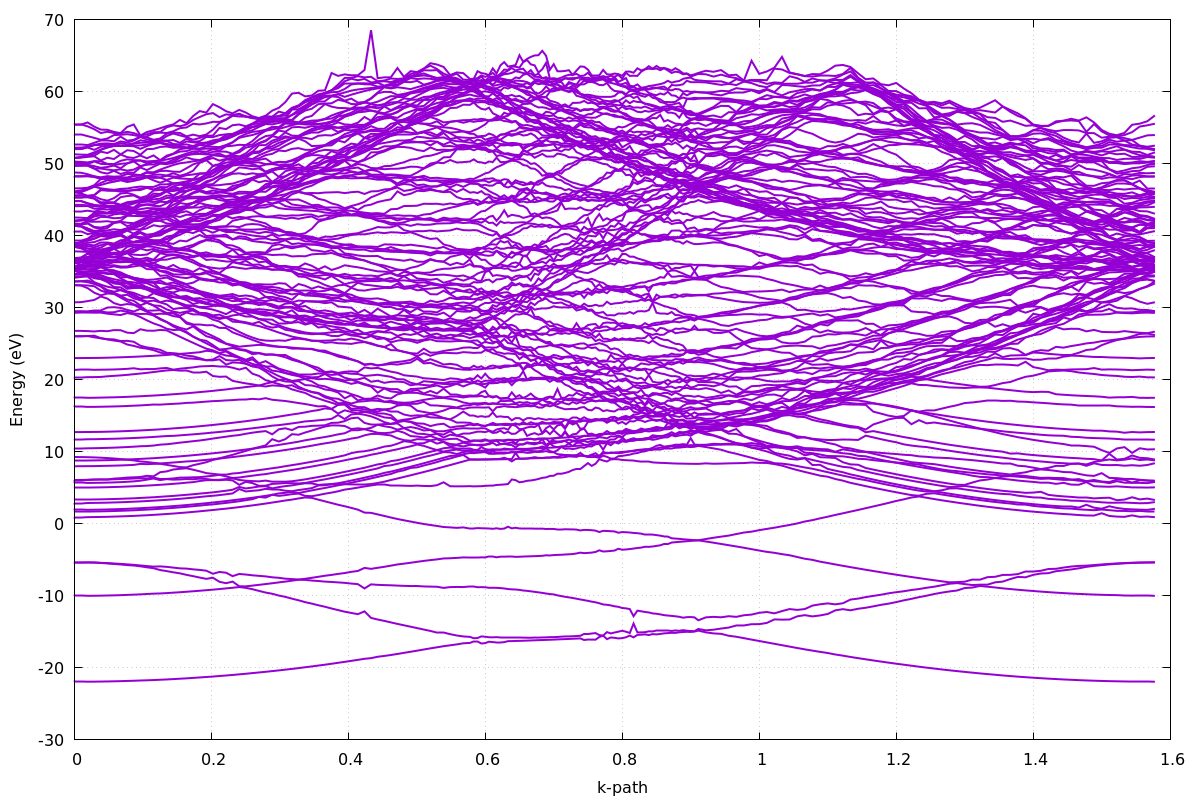
Czwarty krok: Wizualizacja struktury pasmowej (gnuplot)
Ostatnim etapem obliczeń jest wizualizacja struktury pasmowej grafenu na podstawie danych wygenerowanych przez program bands.x. Wynikiem poprzedniego kroku są pliki gr_bands.dat oraz gr_bands.dat.gnu, zawierające energie pasm wzdłuż zadanej ścieżki k.
Utwórz plik plot_bands.gnu o następującej zawartości:
set terminal pngcairo size 1200,800 set output 'gr_bands.png' set xlabel 'k-path' set ylabel 'Energy (eV)' unset key set grid plot 'gr_bands.dat.gnu' using 1:2 with lines lw 2
Następnie uruchom:
gnuplot plot_bands.gnu
Powstanie plik gr_bands.png z wykresem struktury pasmowej grafenu wzdłuż zadanej ścieżki k.
{kind=link}
Uwagi dotyczące skryptów wsadowych
Wszystkie skrypty wsadowe w tym tutorialu rozpoczynają się od:
set -euo pipefailZwiększa to niezawodność i przewidywalność obliczeń na klastrze.
-e(exit on error)
Skrypt natychmiast kończy działanie, jeśli jakiekolwiek polecenie zakończy się błędem. Zapobiega to uruchamianiu kolejnych etapów (np. BANDS lub bands.x) na niekompletnych lub błędnych danych.-u(undefined variables)
Użycie niezdefiniowanej zmiennej powoduje błąd. Chroni to przed literówkami oraz przypadkowym zapisem danych do niewłaściwych katalogów (np./lub/dev/shm).pipefail
W przypadku potoków poleceń (|) błąd dowolnego elementu jest poprawnie wykrywany. Jest to szczególnie istotne przy przekierowaniach i logowaniu wyjścia programów takich jakpw.xczybands.x.